不同参考基因组对RAD-seq分析中SNP Calling的影响
随着新一代测序技术兴起,基于酶切的简化基因组测序(Restriction-site associated DNA sequencing,RAD-seq)由于不受参考基因组的限制,成为快速获取生物高密度单核苷酸多态性(single nucleotide polymorphism,SNP)的主流手段,被广泛运用于变异检测、遗传图谱构建、功能基因挖掘、群体进化分析等研究中。尽管在RAD-seq分析中对SNP Calling方法的评估屡见不鲜,但在分析使用近缘种与物种本种作为参考基因组在SNP Calling上的差异关注不足,尤其是近年来随着全基因组测序物种的日益增加,深入探讨不同参考基因组对SNP Calling的影响将为相关研究提供非常有价值的见解。
中国科学院西双版纳热带植物园(以下简称“版纳植物园”)植物系统发育与多样性保护研究组选取黄杞(Engelhardia roxburghiana)为研究物种,利用生物信息学软件STACKS,探索使用不同参考基因组对SNP Calling的影响。该研究围绕两种不同参考基因组:使用近缘种(即枫杨:Pterocarya stenoptera)作为参考基因组,以及利用物种本种(即黄杞)作为参考基因组。研究发现在使用物种本种作为参考基因组与使用近缘种作为参考基因组之间获得的SNP数量存在显著差异,即本种作为参考基因组产生的SNP数量明显多于使用近缘种作为参考基因组的情形。这一结果表明,选择物种本种的参考基因组为SNP Calling的最优方案;若物种本种参考基因组不可用,则可考虑使用近缘种的参考基因组,同时建议避免选用亲缘关系较远的物种。此项研究强调了参考基因组选择对SNP Calling的影响,丰富了我们对RAD-seq数据分析的理解。
相关结果以Different reference genomes determine different results: Comparing SNP calling in RAD-seq ofEngelhardia roxburghiana using different reference genomes 为题,发表于Plant Science期刊。版纳植物园植物系统发育与多样性保护研究组2022级硕士研究生黄佩涵为该论文第一作者,孟宏虎副研究员、李捷研究员和上海辰山植物园的宋以刚副研究员为该论文通讯作者。该研究得到了国家自然科学基金、中国科学院东南亚中心项目、上海市园林绿化市容科学研究专项基金、中国科学院西部学者项目、云南省兴滇英才支持计划项目以及版纳植物园“十四五”项目的支持。
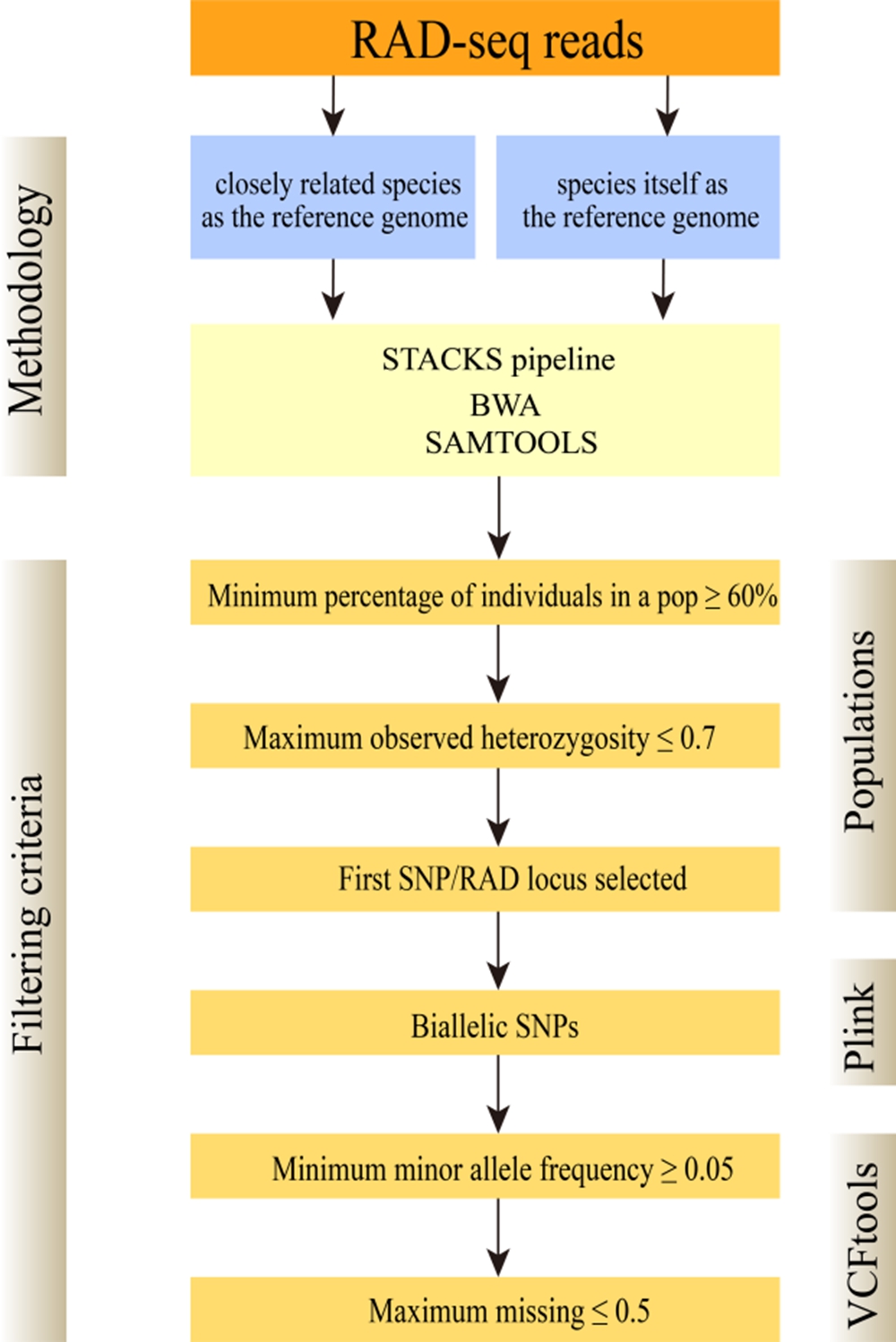
图1研究分析方法流程
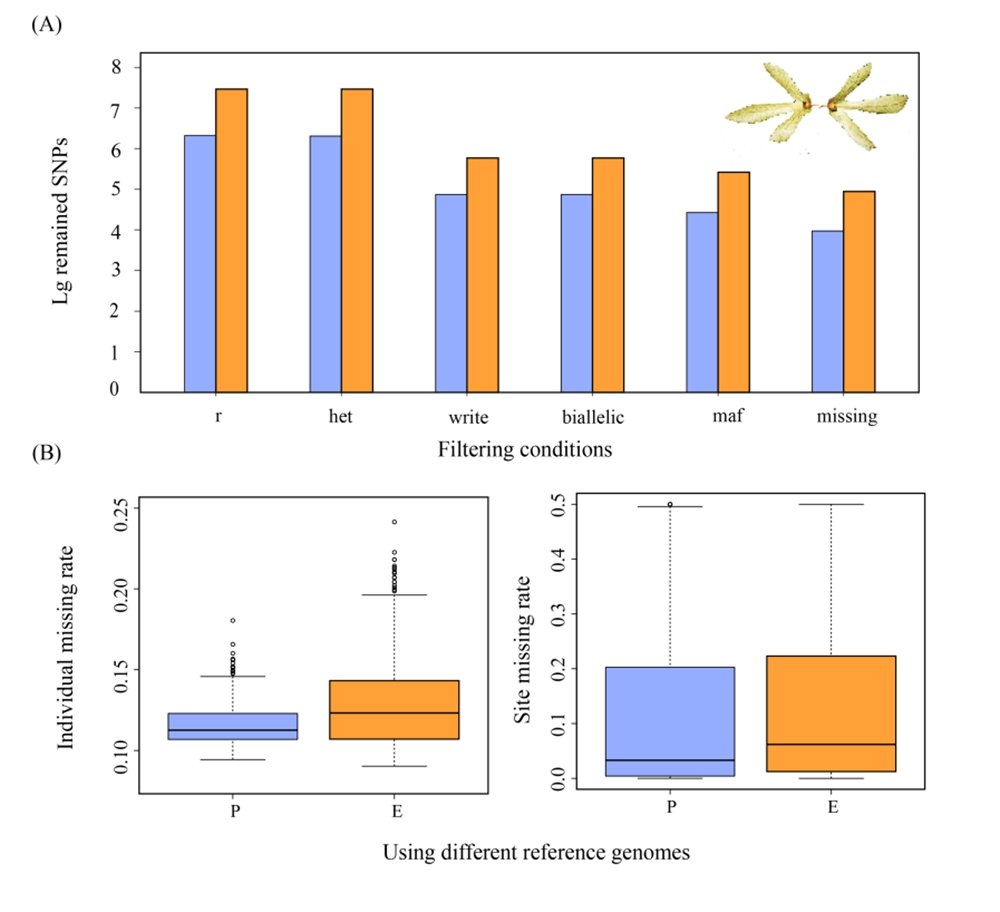
图2 过滤步骤后保留SNP数量的结果,以及保留SNP的位点缺失率和个体缺失率。(A)未配对的括号或引号!不同参考基因组通过过滤步骤从初始到最终的SNP数量的lg转换值。 (B)使用不同参考基因组,获取的最终SNP的位点缺失率和个体缺失率(P:枫杨作为参考基因组;E:黄杞作为参考基因组)